阿爾茨海默病是癡呆的主要原因,其特征是神經病理學的變化,如澱粉樣斑塊、突觸和神經元變性以及神經原纖維纏結。阿爾茨海默病患者大腦中β-澱粉樣蛋白的累積是由於β-澱粉樣蛋白生成和排泄率之間的不平衡造成的。既往研究表明,β-澱粉樣蛋白在腦內的異常積累最終可導致認知功能下降[1-3]。盡管澱粉樣斑塊是阿爾茨海默病在中樞神經系統和外周器官中的重要特征,但靶向清除中樞神經系統中的β-澱粉樣蛋白在阿爾茨海默病治療中療效有限。
肝臟可透過代謝和清除約60%的β-澱粉樣蛋白[4],因而在β-澱粉樣蛋白代謝中發揮核心作用。肝臟可透過肝細胞介導的降解或調節血漿攜帶蛋白水平直接清除β-澱粉樣蛋白,或者調控膽固醇代謝的上下遊間接清除β-澱粉樣蛋白,這種清除代謝在肝臟中受損可能會導致β-澱粉樣蛋白在腦內的累積[1, 5]。值得註意的是,肝臟中膽固醇代謝受損可能會加劇阿爾茨海默病的發展。然而,肝中的膽固醇代謝在β-澱粉樣蛋白清除中的作用仍缺乏深入而全面的總結。
最近來自中國中南大學湘雅二醫院彭偉軍團隊在【中國神經再生研究(英文版)】(Neural Regeneration Research)上發表了題為「 Liver as a new target organ in Alzheimer’s disease: insight from cholesterol metabolism and its role in amyloid-beta clearance 」的綜述。該綜述系統地回顧了阿爾茨海默病的潛在病因,並闡明了肝臟膽固醇代謝在β-澱粉樣蛋白清除中的作用。
β-澱粉樣蛋白的正常代謝和維持β-澱粉樣蛋白產生和清除之間的內穩態平衡對維持大腦健康至關重要。事實上,β-澱粉樣蛋白的生理代謝不僅發生在大腦中,而且也發生在外周。根據外周匯治療策略,大腦和外周組織中的β-澱粉樣蛋白池之間存在動態平衡,肝臟等外周器官在β-澱粉樣蛋白清除中發揮作用[6]。大量的腦源性β-澱粉樣蛋白可透過血腦屏障等途徑進入外周池( 圖1 )。動脈血中的腦源性β-澱粉樣蛋白在穿過周圍器官和組織的毛細血管網(尤其是肝臟)時經歷生理清除[5]。此外,外周β-澱粉樣蛋白分解代謝速率似乎影響β-澱粉樣蛋白從腦內外流的速率,外周來源的β-澱粉樣蛋白可以進入腦內並加速腦內阿爾茨海默病病理進展[7, 8],提示中樞和外周β-澱粉樣蛋白池相互作用和影響。越來越多的研究人員正在關註建立基於外周β-澱粉樣蛋白清除的最佳療法,以建立安全有效的阿爾茨海默病治療方案[9-11]。因此,彭偉軍等認為外周β-澱粉樣蛋白池不僅與阿爾茨海默病相關,而且與阿爾茨海默病有因果關系。

圖1 腦內膽固醇代謝和β-澱粉樣蛋白清除到外周。在大腦中,膽固醇在星形膠質細胞和神經元之間轉移。由星形膠質細胞產生的含膽固醇的脂蛋白顆粒在被鄰近神經元吸收之前被ABCA1轉運蛋白脂化和轉運。載脂蛋白e主要由星形膠質細胞分泌,其功能是轉運腦內膽固醇,並啟動高密度脂蛋白的形成以在細胞間分布脂質。β-澱粉樣蛋白可與載脂蛋白e結合,形成載脂蛋白e-β-澱粉樣蛋白復合物,隨膽固醇排出大腦。神經元透過低密度脂蛋白受體相關蛋白和低密度脂蛋白受體相關蛋白1受體攝取脂蛋白顆粒。神經元將膽固醇轉化為24OHC, 24OHC可以穿過血腦屏障,從而清除大腦中多余的膽固醇(圖源:Wu et al., Neural Regen Res, 2025)
迴圈中的β-澱粉樣蛋白主要透過肝細胞內降解或膽汁內直接排泄的方式被清除。低密度脂蛋白受體相關蛋白1被認為是與迴圈β-澱粉樣蛋白結合的肝臟重要受體[12-14],低密度脂蛋白受體相關蛋白1在肝竇內皮細胞中高表達[15]。而肝功能異常可能導致肝臟低密度脂蛋白受體相關蛋白1表達低下和肝細胞直接從血液中攝取和降解β-澱粉樣蛋白的減少( 圖2 )[16]。肝功能障礙也可能間接導致β-澱粉樣蛋白相關蛋白和脂質代謝失調,從而損害β-澱粉樣蛋白轉運和清除。
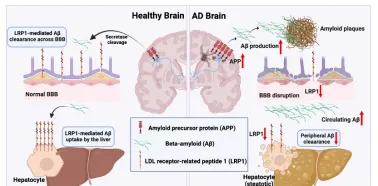
圖2 低密度脂蛋白受體相關蛋白1是肝臟飽和攝取血漿遊離β-澱粉樣蛋白的重要受體。目前認為,中樞和外周β-澱粉樣蛋白清除(分別在血腦屏障和肝臟)的主要機制是由低密度脂蛋白受體相關蛋白1介導。肝功能障礙可顯著降低低密度脂蛋白受體相關蛋白1水平,導致澱粉樣蛋白負荷水平升高和阿爾茨海默病的發生,這些特征可能會加劇血腦屏障功能障礙(圖源:Wu et al., Neural Regen Res, 2025)
肝臟膽固醇代謝被認為是阿爾茨海默病進展的關鍵環節。研究表明,膽固醇外流可減少β-澱粉樣蛋白積累,因為β分泌酶會在膽固醇大量存在時對APP進行初始裂解[17],這表明保持大腦中最佳的膽固醇水平可能是降低阿爾茨海默病進展風險的一個重要因素。75%由肝臟內源性產生[18],肝臟也是唯一能夠透過分泌到膽汁並將其轉化為膽汁酸來清除多余膽固醇的器官,這對維持機體膽固醇穩態至關重要。高膽固醇血癥被認為是阿爾茨海默病的危險因素[17, 19-21]。膽固醇穩態相關蛋白表達的改變與阿爾茨海默病的發生或發展有關,這可能依賴於高膽固醇血癥下血腦屏障通透性的增強[22]。
膽汁酸由肝臟產生,並作為膽固醇代謝和消除的副產物儲存在膽囊中。膽汁酸透過其高表達的受體低密度脂蛋白受體相關蛋白1和低密度脂蛋白受體相關蛋白介導的肝細胞攝取在迴圈β-澱粉樣蛋白的清除中起著至關重要的作用[23]。膽汁中β-澱粉樣蛋白排泄減少可能與肝功能障礙有關。某些內源性膽汁酸,尤其是牛磺熊去氧膽酸,具有透過血腦屏障的能力,被認為是強大的神經保護劑和改善阿爾茨海默病的潛在療法[24]。研究發現,與對照小鼠相比,APP/PS1小鼠中的牛磺熊去氧膽酸水平顯著較低[25]。牛磺熊去氧膽酸可改善神經退行性疾病模型的學習記憶能力和β-澱粉樣蛋白神經病理改變( 圖3 )[26]。
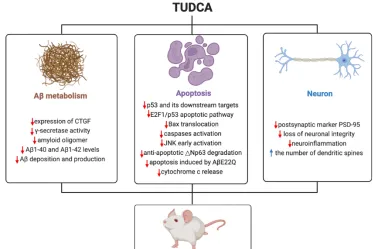
圖3 牛磺熊去氧膽酸在阿爾茨海默病實驗模型中具有有益作用。在實驗性阿爾茨海默病模型中,牛磺熊去氧膽酸透過抑制多種促雕亡通路和促進抗雕亡過程來阻斷雕亡。此外,牛磺熊去氧膽酸減少了突觸遺失和β-澱粉樣蛋白肽在額葉皮質和海馬的積累。在阿爾茨海默病小鼠中,每一種情況都有助於增加空間、辨識和上下文記憶(圖源:Wu et al., Neural Regen Res, 2025)
此外,在肝臟中合成的三甲胺N-氧化物水平升高加重了神經退行性過程,從而促進了阿爾茨海默病的發病[27, 28]。三甲胺N-氧化物水平與β-澱粉樣蛋白斑塊沈積增加之間存在正相關[29]。三甲胺N-氧化物可能對膽固醇逆向轉運有不利影響[30],也可以抑制膽汁酸的合成[31]。在超生理濃度下,三甲胺N-氧化物可加速β-澱粉樣蛋白原纖維向β-澱粉樣蛋白折疊構象的轉變,這是原纖維生成所需的[32]。這些纖維有聚集形成斑塊的趨勢,提示三甲胺N-氧化物可能透過促進β-澱粉樣蛋白聚集形成斑塊在阿爾茨海默病發病機制中發揮作用。但目前關於三甲胺N-氧化物的作用的研究結論並不一致,彭偉軍等認為可能與其濃度有關。在生理濃度下,三甲胺N-氧化物往往具有積極作用,如改善血腦屏障完整性。然而,超過生理濃度的三甲胺N-氧化物往往具有負面影響[30]( 圖4 )。
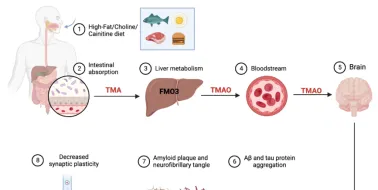
圖4 三甲胺N-氧化物對阿爾茨海默病病理生理學的潛在貢獻。膳食成分(如高脂、膽堿和肉堿)被腸道細菌發酵,然後被腸道中的TMA-解離酶轉化為TMA,並在肝臟中被FMO3氧化合成三甲胺N-氧化物。三甲胺N-氧化物可透過BBB到達中樞神經系統,導致β-澱粉樣蛋白和tau蛋白聚集,降低突觸可塑性(圖源:Wu et al., Neural Regen Res, 2025)
總之,彭偉軍等綜述了肝臟是外周參與β-澱粉樣蛋白代謝的主要器官,在阿爾茨海默病的病理生理中起著至關重要的作用。恢復肝臟正常的膽固醇代謝可能是治療阿爾茨海默病的一個有前景的治療策略。以肝臟為靶點減少β-澱粉樣蛋白生成或增加外周組織清除可能是開發改善癡呆的有效新藥的一種新的潛在治療方法。
當然該綜述也存在一定性局限性。首先,目前的研究對導致阿爾茨海默病發展的肝臟病理的具體類別和嚴重程度仍然知之甚少;其次,該綜述中參照的大多數研究來自使用細胞和動物模型的臨床前研究,而臨床試驗較少。未來需要進一步的研究來充分了解肝臟在阿爾茨海默病中的具體作用和潛力。
原文連結: https://doi.org/10.4103/1673-5374.391305
參考文獻
[1] Cheng Y, Tian DY, Wang YJ. Peripheral clearance of brain-derived Aβ in Alzheimer's disease: pathophysiology and therapeutic perspectives. Transl Neurodegener. 2020;9(1):16.
[2] Yuan P, Zhang M, Tong L, et al. PLD3 affects axonal spheroids and network defects in Alzheimer's disease. Nature. 2022;612(7939):328-337.
[3] Wang J, Mei Y, Zhang X, et al. Aberrant serotonergic signaling contributes to the hyperexcitability of CA1 pyramidal neurons in a mouse model of Alzheimer's disease. Cell Rep. 2023;42(3):112152.
[4] Wang J, Gu BJ, Masters CL, et al. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612-623.
[5] Xiang Y, Bu XL, Liu YH, et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 2015;130(4):487-499.
[6] Georgievska B, Gustavsson S, Lundkvist J, et al. Revisiting the peripheral sink hypothesis: inhibiting BACE1 activity in the periphery does not alter β-amyloid levels in the CNS. J Neurochem. 2015;132(4):477-486.
[7] Kauwe G, Tracy TE. Amyloid beta emerges from below the neck to disable the brain. PLoS Biol. 2021;19(9):e3001388.
[8] Bu XL, Xiang Y, Jin WS, et al. Blood-derived amyloid-β protein induces Alzheimer's disease pathologies. Mol Psychiatry. 2018;23(9):1948-1956.
[9] Wu L, Jiang W, Zhao N, et al. Heparan sulfate from porcine mucosa promotes amyloid-beta clearance in APP/PS1 mice and alleviates Alzheimer's pathology. Carbohydr Polym. 2022;285:119205.
[10] Xu L, Li L, Pan CL, et al. Erythropoietin signaling in peripheral macrophages is required for systemic β-amyloid clearance. EMBO J. 2022;41(22):e111038.
[11] Liu X, Che R, Liang W, et al. Clusterin transduces Alzheimer-risk signals to amyloidogenesis. Signal Transduct Target Ther. 2022;7(1):325.
[12] Mohamed LA, Kaddoumi A. In vitro investigation of amyloid-β hepatobiliary disposition in sandwich-cultured primary rat hepatocytes. Drug Metab Dispos. 2013;41(10):1787-1796.
[13] Yamagishi S, Matsui T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur J Med Res. 2015;20(1):15.
[14] Bassendine MF, Taylor-Robinson SD, Fertleman M, et al. Is Alzheimer's disease a liver disease of the brain? J Alzheimers Dis. 2020;75(1):1-14.
[15] Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front Aging Neurosci. 2014;6:93.
[16] Garcia J, Chang R, Steinberg RA, et al. Modulation of hepatic amyloid precursor protein and lipoprotein receptor-related protein 1 by chronic alcohol intake: potential link between liver steatosis and amyloid-β. Front Physiol. 2022;13:930402.
[17] Roca-Agujetas V, Barbero-Camps E, De Dios C, et al. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer's disease. Mol Neurodegener. 2021;16(1):15.
[18] Kapourchali FR, Surendiran G, Goulet A, et al. The role of dietary cholesterol in lipoprotein metabolism and related metabolic abnormalities: a mini-review. Crit Rev Food Sci Nutr. 2016;56(14):2408-2415.
[19] Andrews SJ, Fulton-Howard B, O'reilly P, et al. Causal associations between modifiable risk factors and the Alzheimer's phenome. Ann Neurol. 2021;89(1):54-65.
[20] Dai L, Zou L, Meng L, et al. Cholesterol metabolism in neurodegenerative diseases: molecular mechanisms and therapeutic targets. Mol Neurobiol. 2021;58(5):2183-2201.
[21] Maiuolo J, Gliozzi M, Musolino V, et al. The role of endothelial dysfunction in peripheral blood nerve barrier: molecular mechanisms and pathophysiological implications. Int J Mol Sci. 2019;20(12):3022.
[22] Chen X, Gawryluk JW, Wagener JF, et al. Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer's disease. J Neuroinflammation. 2008;5:12.
[23] Kanekiyo T, Cirrito JR, Liu CC, et al. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci. 2013;33(49):19276-19283.
[24] Pan X, Elliott CT, Mcguinness B, et al. Metabolomic profiling of bile acids in clinical and experimental samples of Alzheimer's disease. Metabolites. 2017;7(2):28.
[25] Kaur H, Seeger D, Golovko S, et al. Liver bile acid changes in mouse models of Alzheimer's disease. Int J Mol Sci. 2021;22(14):7451.
[26] Ochiai T, Nagayama T, Matsui K, et al. Tauroursodeoxycholic acid attenuates diet-induced and age-related peripheral endoplasmic reticulum stress and cerebral amyloid pathology in a mouse model of Alzheimer's disease. J Prev Alzheimers Dis. 2021;8(4):483-494.
[27] Gao Q, Wang Y, Wang X, et al. Decreased levels of circulating trimethylamine N-oxide alleviate cognitive and pathological deterioration in transgenic mice: a potential therapeutic approach for Alzheimer's disease. Aging (Albany NY). 2019;11(19):8642-8663.
[28] Govindarajulu M, Pinky PD, Steinke I, et al. Gut metabolite TMAO induces synaptic plasticity deficits by promoting endoplasmic reticulum stress. Front Mol Neurosci. 2020;13:138.
[29] Li D, Ke Y, Zhan R, et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell. 2018;17(4):e12768.
[30] Zhang L, Yu F, Xia J. Trimethylamine N-oxide: role in cell senescence and age-related diseases. Eur J Nutr. 2023;62(2):525-541.
[31] Ding L, Chang M, Guo Y, et al. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018;17(1):286.
[32] Kumari A, Rajput R, Shrivastava N, et al. Synergistic approaches unraveling regulation and aggregation of intrinsically disordered β-amyloids implicated in Alzheimer's disease. Int J Biochem Cell Biol. 2018;99:19-27.
文章摘要: 阿爾茨海默病是癡呆的主要原因,其特征是神經病理學的變化,如澱粉樣斑塊、突觸和神經元變性以及神經原纖維纏結。盡管澱粉樣斑塊是阿爾茨海默病在中樞神經系統和外周器官中的重要特征,但靶向清除中樞神經系統中的β-澱粉樣蛋白在阿爾茨海默病治療中療效有限。阿爾茨海默病患者常見代謝異常。肝臟是參與β-澱粉樣蛋白代謝的主要外周器官,在阿爾茨海默病的病理生理方面中起著至關重要的作用。值得註意的是,肝臟中膽固醇代謝受損可能會加劇阿爾茨海默病的發展。因此此次綜述系統地回顧了阿爾茨海默病的潛在病因,並闡明了肝臟膽固醇代謝在β-澱粉樣蛋白清除中的作用。文章提出,恢復肝臟中正常的膽固醇代謝可能是治療阿爾茨海默病的一種潛在治療策略。
文章關鍵詞: 阿爾茨海默病;β-澱粉樣蛋白;外周清除;肝臟;低密度脂蛋白受體相關蛋白1;載脂蛋白e;ABCA1;肝臟X受體;膽固醇代謝;膽汁酸;牛磺熊去氧膽酸;三甲胺N-氧化物
文章來源: Wu B, Liu Y, Li H, Zhu L, Zeng L, Zhang Z, Peng W (2025) Liver as a new target organ in Alzheimer’s disease: insight from cholesterol metabolism and its role in amyloid-beta clearance. Neural Regen Res 20(3):695-714.











