阿尔茨海默病是痴呆的主要原因,其特征是神经病理学的变化,如淀粉样斑块、突触和神经元变性以及神经原纤维缠结。阿尔茨海默病患者大脑中β-淀粉样蛋白的累积是由于β-淀粉样蛋白生成和排泄率之间的不平衡造成的。既往研究表明,β-淀粉样蛋白在脑内的异常积累最终可导致认知功能下降[1-3]。尽管淀粉样斑块是阿尔茨海默病在中枢神经系统和外周器官中的重要特征,但靶向清除中枢神经系统中的β-淀粉样蛋白在阿尔茨海默病治疗中疗效有限。
肝脏可通过代谢和清除约60%的β-淀粉样蛋白[4],因而在β-淀粉样蛋白代谢中发挥核心作用。肝脏可通过肝细胞介导的降解或调节血浆载体蛋白水平直接清除β-淀粉样蛋白,或者调控胆固醇代谢的上下游间接清除β-淀粉样蛋白,这种清除代谢在肝脏中受损可能会导致β-淀粉样蛋白在脑内的累积[1, 5]。值得注意的是,肝脏中胆固醇代谢受损可能会加剧阿尔茨海默病的发展。然而,肝中的胆固醇代谢在β-淀粉样蛋白清除中的作用仍缺乏深入而全面的总结。
最近来自中国中南大学湘雅二医院彭伟军团队在【中国神经再生研究(英文版)】(Neural Regeneration Research)上发表了题为「 Liver as a new target organ in Alzheimer’s disease: insight from cholesterol metabolism and its role in amyloid-beta clearance 」的综述。该综述系统地回顾了阿尔茨海默病的潜在病因,并阐明了肝脏胆固醇代谢在β-淀粉样蛋白清除中的作用。
β-淀粉样蛋白的正常代谢和维持β-淀粉样蛋白产生和清除之间的内稳态平衡对维持大脑健康至关重要。事实上,β-淀粉样蛋白的生理代谢不仅发生在大脑中,而且也发生在外周。根据外周汇治疗策略,大脑和外周组织中的β-淀粉样蛋白池之间存在动态平衡,肝脏等外周器官在β-淀粉样蛋白清除中发挥作用[6]。大量的脑源性β-淀粉样蛋白可通过血脑屏障等途径进入外周池( 图1 )。动脉血中的脑源性β-淀粉样蛋白在穿过周围器官和组织的毛细血管网(尤其是肝脏)时经历生理清除[5]。此外,外周β-淀粉样蛋白分解代谢速率似乎影响β-淀粉样蛋白从脑内外流的速率,外周来源的β-淀粉样蛋白可以进入脑内并加速脑内阿尔茨海默病病理进展[7, 8],提示中枢和外周β-淀粉样蛋白池相互作用和影响。越来越多的研究人员正在关注建立基于外周β-淀粉样蛋白清除的最佳疗法,以创建安全有效的阿尔茨海默病治疗方案[9-11]。因此,彭伟军等认为外周β-淀粉样蛋白池不仅与阿尔茨海默病相关,而且与阿尔茨海默病有因果关系。

图1 脑内胆固醇代谢和β-淀粉样蛋白清除到外周。在大脑中,胆固醇在星形胶质细胞和神经元之间转移。由星形胶质细胞产生的含胆固醇的脂蛋白颗粒在被邻近神经元吸收之前被ABCA1转运蛋白脂化和转运。载脂蛋白e主要由星形胶质细胞分泌,其功能是转运脑内胆固醇,并启动高密度脂蛋白的形成以在细胞间分布脂质。β-淀粉样蛋白可与载脂蛋白e结合,形成载脂蛋白e-β-淀粉样蛋白复合物,随胆固醇排出大脑。神经元通过低密度脂蛋白受体相关蛋白和低密度脂蛋白受体相关蛋白1受体摄取脂蛋白颗粒。神经元将胆固醇转化为24OHC, 24OHC可以穿过血脑屏障,从而清除大脑中多余的胆固醇(图源:Wu et al., Neural Regen Res, 2025)
循环中的β-淀粉样蛋白主要通过肝细胞内降解或胆汁内直接排泄的方式被清除。低密度脂蛋白受体相关蛋白1被认为是与循环β-淀粉样蛋白结合的肝脏重要受体[12-14],低密度脂蛋白受体相关蛋白1在肝窦内皮细胞中高表达[15]。而肝功能异常可能导致肝脏低密度脂蛋白受体相关蛋白1表达低下和肝细胞直接从血液中摄取和降解β-淀粉样蛋白的减少( 图2 )[16]。肝功能障碍也可能间接导致β-淀粉样蛋白相关蛋白和脂质代谢失调,从而损害β-淀粉样蛋白转运和清除。
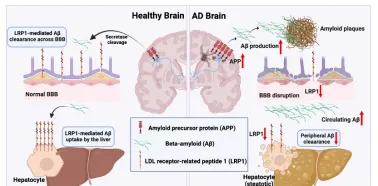
图2 低密度脂蛋白受体相关蛋白1是肝脏饱和摄取血浆游离β-淀粉样蛋白的重要受体。目前认为,中枢和外周β-淀粉样蛋白清除(分别在血脑屏障和肝脏)的主要机制是由低密度脂蛋白受体相关蛋白1介导。肝功能障碍可显著降低低密度脂蛋白受体相关蛋白1水平,导致淀粉样蛋白负荷水平升高和阿尔茨海默病的发生,这些特征可能会加剧血脑屏障功能障碍(图源:Wu et al., Neural Regen Res, 2025)
肝脏胆固醇代谢被认为是阿尔茨海默病进展的关键环节。研究表明,胆固醇外流可减少β-淀粉样蛋白积累,因为β分泌酶会在胆固醇大量存在时对APP进行初始裂解[17],这表明保持大脑中最佳的胆固醇水平可能是降低阿尔茨海默病进展风险的一个重要因素。75%由肝脏内源性产生[18],肝脏也是唯一能够通过分泌到胆汁并将其转化为胆汁酸来清除多余胆固醇的器官,这对维持机体胆固醇稳态至关重要。高胆固醇血症被认为是阿尔茨海默病的危险因素[17, 19-21]。胆固醇稳态相关蛋白表达的改变与阿尔茨海默病的发生或发展有关,这可能依赖于高胆固醇血症下血脑屏障通透性的增强[22]。
胆汁酸由肝脏产生,并作为胆固醇代谢和消除的副产物储存在胆囊中。胆汁酸通过其高表达的受体低密度脂蛋白受体相关蛋白1和低密度脂蛋白受体相关蛋白介导的肝细胞摄取在循环β-淀粉样蛋白的清除中起着至关重要的作用[23]。胆汁中β-淀粉样蛋白排泄减少可能与肝功能障碍有关。某些内源性胆汁酸,尤其是牛磺熊去氧胆酸,具有通过血脑屏障的能力,被认为是强大的神经保护剂和改善阿尔茨海默病的潜在疗法[24]。研究发现,与对照小鼠相比,APP/PS1小鼠中的牛磺熊去氧胆酸水平显著较低[25]。牛磺熊去氧胆酸可改善神经退行性疾病模型的学习记忆能力和β-淀粉样蛋白神经病理改变( 图3 )[26]。
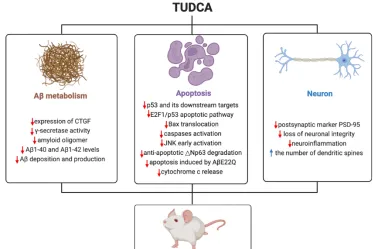
图3 牛磺熊去氧胆酸在阿尔茨海默病实验模型中具有有益作用。在实验性阿尔茨海默病模型中,牛磺熊去氧胆酸通过抑制多种促凋亡通路和促进抗凋亡过程来阻断凋亡。此外,牛磺熊去氧胆酸减少了突触丢失和β-淀粉样蛋白肽在额叶皮质和海马的积累。在阿尔茨海默病小鼠中,每一种情况都有助于增加空间、识别和上下文记忆(图源:Wu et al., Neural Regen Res, 2025)
此外,在肝脏中合成的三甲胺N-氧化物水平升高加重了神经退行性过程,从而促进了阿尔茨海默病的发病[27, 28]。三甲胺N-氧化物水平与β-淀粉样蛋白斑块沉积增加之间存在正相关[29]。三甲胺N-氧化物可能对胆固醇逆向转运有不利影响[30],也可以抑制胆汁酸的合成[31]。在超生理浓度下,三甲胺N-氧化物可加速β-淀粉样蛋白原纤维向β-淀粉样蛋白折叠构象的转变,这是原纤维生成所需的[32]。这些纤维有聚集形成斑块的趋势,提示三甲胺N-氧化物可能通过促进β-淀粉样蛋白聚集形成斑块在阿尔茨海默病发病机制中发挥作用。但目前关于三甲胺N-氧化物的作用的研究结论并不一致,彭伟军等认为可能与其浓度有关。在生理浓度下,三甲胺N-氧化物往往具有积极作用,如改善血脑屏障完整性。然而,超过生理浓度的三甲胺N-氧化物往往具有负面影响[30]( 图4 )。
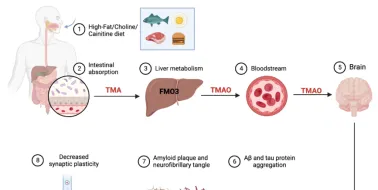
图4 三甲胺N-氧化物对阿尔茨海默病病理生理学的潜在贡献。膳食成分(如高脂、胆碱和肉碱)被肠道细菌发酵,然后被肠道中的TMA-裂合酶转化为TMA,并在肝脏中被FMO3氧化合成三甲胺N-氧化物。三甲胺N-氧化物可通过BBB到达中枢神经系统,导致β-淀粉样蛋白和tau蛋白聚集,降低突触可塑性(图源:Wu et al., Neural Regen Res, 2025)
总之,彭伟军等综述了肝脏是外周参与β-淀粉样蛋白代谢的主要器官,在阿尔茨海默病的病理生理中起着至关重要的作用。恢复肝脏正常的胆固醇代谢可能是治疗阿尔茨海默病的一个有前景的治疗策略。以肝脏为靶点减少β-淀粉样蛋白生成或增加外周组织清除可能是开发改善痴呆的有效新药的一种新的潜在治疗方法。
当然该综述也存在一定性局限性。首先,目前的研究对导致阿尔茨海默病发展的肝脏病理的具体类型和严重程度仍然知之甚少;其次,该综述中引用的大多数研究来自使用细胞和动物模型的临床前研究,而临床试验较少。未来需要进一步的研究来充分了解肝脏在阿尔茨海默病中的具体作用和潜力。
原文链接: https://doi.org/10.4103/1673-5374.391305
参考文献
[1] Cheng Y, Tian DY, Wang YJ. Peripheral clearance of brain-derived Aβ in Alzheimer's disease: pathophysiology and therapeutic perspectives. Transl Neurodegener. 2020;9(1):16.
[2] Yuan P, Zhang M, Tong L, et al. PLD3 affects axonal spheroids and network defects in Alzheimer's disease. Nature. 2022;612(7939):328-337.
[3] Wang J, Mei Y, Zhang X, et al. Aberrant serotonergic signaling contributes to the hyperexcitability of CA1 pyramidal neurons in a mouse model of Alzheimer's disease. Cell Rep. 2023;42(3):112152.
[4] Wang J, Gu BJ, Masters CL, et al. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612-623.
[5] Xiang Y, Bu XL, Liu YH, et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 2015;130(4):487-499.
[6] Georgievska B, Gustavsson S, Lundkvist J, et al. Revisiting the peripheral sink hypothesis: inhibiting BACE1 activity in the periphery does not alter β-amyloid levels in the CNS. J Neurochem. 2015;132(4):477-486.
[7] Kauwe G, Tracy TE. Amyloid beta emerges from below the neck to disable the brain. PLoS Biol. 2021;19(9):e3001388.
[8] Bu XL, Xiang Y, Jin WS, et al. Blood-derived amyloid-β protein induces Alzheimer's disease pathologies. Mol Psychiatry. 2018;23(9):1948-1956.
[9] Wu L, Jiang W, Zhao N, et al. Heparan sulfate from porcine mucosa promotes amyloid-beta clearance in APP/PS1 mice and alleviates Alzheimer's pathology. Carbohydr Polym. 2022;285:119205.
[10] Xu L, Li L, Pan CL, et al. Erythropoietin signaling in peripheral macrophages is required for systemic β-amyloid clearance. EMBO J. 2022;41(22):e111038.
[11] Liu X, Che R, Liang W, et al. Clusterin transduces Alzheimer-risk signals to amyloidogenesis. Signal Transduct Target Ther. 2022;7(1):325.
[12] Mohamed LA, Kaddoumi A. In vitro investigation of amyloid-β hepatobiliary disposition in sandwich-cultured primary rat hepatocytes. Drug Metab Dispos. 2013;41(10):1787-1796.
[13] Yamagishi S, Matsui T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur J Med Res. 2015;20(1):15.
[14] Bassendine MF, Taylor-Robinson SD, Fertleman M, et al. Is Alzheimer's disease a liver disease of the brain? J Alzheimers Dis. 2020;75(1):1-14.
[15] Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front Aging Neurosci. 2014;6:93.
[16] Garcia J, Chang R, Steinberg RA, et al. Modulation of hepatic amyloid precursor protein and lipoprotein receptor-related protein 1 by chronic alcohol intake: potential link between liver steatosis and amyloid-β. Front Physiol. 2022;13:930402.
[17] Roca-Agujetas V, Barbero-Camps E, De Dios C, et al. Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer's disease. Mol Neurodegener. 2021;16(1):15.
[18] Kapourchali FR, Surendiran G, Goulet A, et al. The role of dietary cholesterol in lipoprotein metabolism and related metabolic abnormalities: a mini-review. Crit Rev Food Sci Nutr. 2016;56(14):2408-2415.
[19] Andrews SJ, Fulton-Howard B, O'reilly P, et al. Causal associations between modifiable risk factors and the Alzheimer's phenome. Ann Neurol. 2021;89(1):54-65.
[20] Dai L, Zou L, Meng L, et al. Cholesterol metabolism in neurodegenerative diseases: molecular mechanisms and therapeutic targets. Mol Neurobiol. 2021;58(5):2183-2201.
[21] Maiuolo J, Gliozzi M, Musolino V, et al. The role of endothelial dysfunction in peripheral blood nerve barrier: molecular mechanisms and pathophysiological implications. Int J Mol Sci. 2019;20(12):3022.
[22] Chen X, Gawryluk JW, Wagener JF, et al. Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer's disease. J Neuroinflammation. 2008;5:12.
[23] Kanekiyo T, Cirrito JR, Liu CC, et al. Neuronal clearance of amyloid-β by endocytic receptor LRP1. J Neurosci. 2013;33(49):19276-19283.
[24] Pan X, Elliott CT, Mcguinness B, et al. Metabolomic profiling of bile acids in clinical and experimental samples of Alzheimer's disease. Metabolites. 2017;7(2):28.
[25] Kaur H, Seeger D, Golovko S, et al. Liver bile acid changes in mouse models of Alzheimer's disease. Int J Mol Sci. 2021;22(14):7451.
[26] Ochiai T, Nagayama T, Matsui K, et al. Tauroursodeoxycholic acid attenuates diet-induced and age-related peripheral endoplasmic reticulum stress and cerebral amyloid pathology in a mouse model of Alzheimer's disease. J Prev Alzheimers Dis. 2021;8(4):483-494.
[27] Gao Q, Wang Y, Wang X, et al. Decreased levels of circulating trimethylamine N-oxide alleviate cognitive and pathological deterioration in transgenic mice: a potential therapeutic approach for Alzheimer's disease. Aging (Albany NY). 2019;11(19):8642-8663.
[28] Govindarajulu M, Pinky PD, Steinke I, et al. Gut metabolite TMAO induces synaptic plasticity deficits by promoting endoplasmic reticulum stress. Front Mol Neurosci. 2020;13:138.
[29] Li D, Ke Y, Zhan R, et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell. 2018;17(4):e12768.
[30] Zhang L, Yu F, Xia J. Trimethylamine N-oxide: role in cell senescence and age-related diseases. Eur J Nutr. 2023;62(2):525-541.
[31] Ding L, Chang M, Guo Y, et al. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018;17(1):286.
[32] Kumari A, Rajput R, Shrivastava N, et al. Synergistic approaches unraveling regulation and aggregation of intrinsically disordered β-amyloids implicated in Alzheimer's disease. Int J Biochem Cell Biol. 2018;99:19-27.
文章摘要: 阿尔茨海默病是痴呆的主要原因,其特征是神经病理学的变化,如淀粉样斑块、突触和神经元变性以及神经原纤维缠结。尽管淀粉样斑块是阿尔茨海默病在中枢神经系统和外周器官中的重要特征,但靶向清除中枢神经系统中的β-淀粉样蛋白在阿尔茨海默病治疗中疗效有限。阿尔茨海默病患者常见代谢异常。肝脏是参与β-淀粉样蛋白代谢的主要外周器官,在阿尔茨海默病的病理生理方面中起着至关重要的作用。值得注意的是,肝脏中胆固醇代谢受损可能会加剧阿尔茨海默病的发展。因此此次综述系统地回顾了阿尔茨海默病的潜在病因,并阐明了肝脏胆固醇代谢在β-淀粉样蛋白清除中的作用。文章提出,恢复肝脏中正常的胆固醇代谢可能是治疗阿尔茨海默病的一种潜在治疗策略。
文章关键词: 阿尔茨海默病;β-淀粉样蛋白;外周清除;肝脏;低密度脂蛋白受体相关蛋白1;载脂蛋白e;ABCA1;肝脏X受体;胆固醇代谢;胆汁酸;牛磺熊去氧胆酸;三甲胺N-氧化物
文章来源: Wu B, Liu Y, Li H, Zhu L, Zeng L, Zhang Z, Peng W (2025) Liver as a new target organ in Alzheimer’s disease: insight from cholesterol metabolism and its role in amyloid-beta clearance. Neural Regen Res 20(3):695-714.











