一、CRC是做什麽的?
CRC(Clinical resarch coordinator)即臨床協調員,是指經主要研究者(PI)授權在臨床試驗中協助研究者進行非醫學判斷的相關事務性工作,是臨床試驗的參與者、協調者。
二、臨床試驗流程
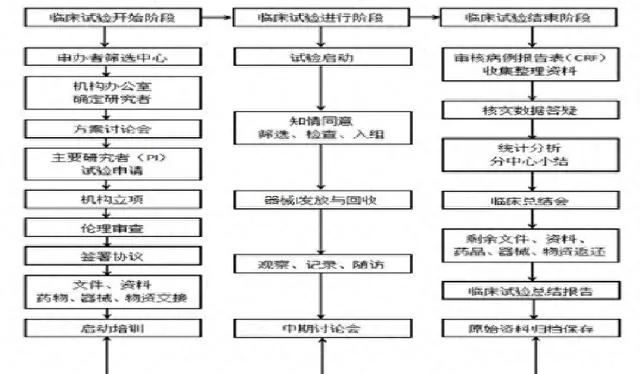
三、臨床試驗的各個階段
1、SSU階段(啟動前)
(1)中心調研
中心調研是申辦方/CRO與研究者/研究中心雙向選擇的過程。申辦方通常開啟專案都會有專案可行性調研。一般申辦方產品對應的適應癥會有合作習慣的主要專家和醫院,於是一般申辦方會有意向中心和研究者清單,甚至合作的牽頭專家自然而然會有團隊(協會、聯盟等),最後根據試驗分類和規模需要更多中心和團隊成員,產生中心調研和篩選的工作。沒有可參考意向中心清單提供的,對於承接業務的CRO/SMO也會有合作關系較好的推薦機構名單。
中心調研內容包括但不限於:推薦科室、推薦研究者、中心幹系人聯系方式(科室、機構、倫理等)、科室器材情況、競爭專案情況、病源量、官方招募渠道、機構立項流程、倫理審查流程、遺傳辦蓋章流程、協定簽署蓋章流程、財務要求、研究產品入庫流程、啟動會要求、中心特殊費用以及特殊要求等等。
(2)機構立項
在機構確認承接專案並且拿到組長單位倫理批件後,根據中心「立項檔清單」協助CRA收集立項資料、完成研究者檔簽字,協助遞交立項資料。大部份機構需要先透過郵箱發送電子版檔給機構秘書形審後再遞交紙質版,具體視機構流程而定。
CRC須了解機構立項大約時長,部份機構會有一些特殊流程,比如是否機構立項前要先召開研究者會,是否有立項會,有可能涉及到的費用問題等,要提前提醒CRA,以免影響專案進度。
(3)倫理審查
倫理遞交流程與機構立項大體相似,大部份中心初始審查需拿到組長單位批件並在機構立項透過後。通常倫理審查的形式有會議審查、快速審查、檔備案。對於初始審查來講一般需要會審,符合區域倫理互認的中心快審,但快審不一定比會審快。
CRC需了解中心倫理會時間、倫理會頻率、本次倫理會截止遞交時間、遞交頻率、遞交至取得首次倫理意見時間、遞交至取得倫理批件的大概時間等。
(4)遺傳辦
遺傳辦:中國人類遺傳資源管理辦公室
向哪裏申報?
國務院科學技術行政主管部門和衛生行政主管部門共同負責管理,並聯合成立遺傳辦,暫設在國務院科學技術行政主管部門。
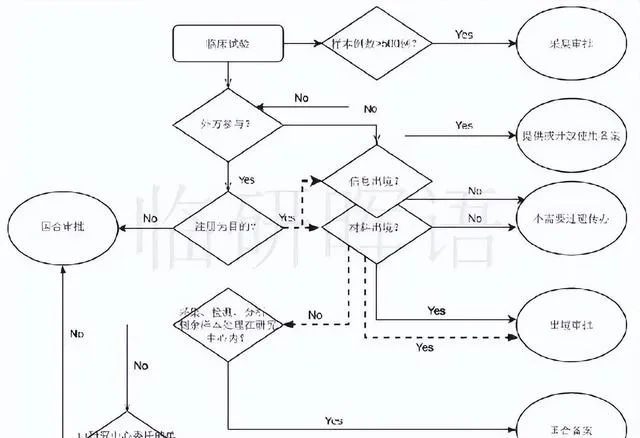
什麽情況下需要遺傳辦申報?
如下圖所示。
人類遺傳資源:包括人類遺傳資源材料和人類遺傳資源資訊;
中國人類遺傳資源采集審批:此項審批適用於在中國境內從事的中國人類遺傳資源采集活動,包括重要遺傳家系、特定地區人類遺傳資源和國務院科學技術行政部門規定種類、數量的人類遺傳資源的采集活動的規範和管理。
國際合作科學研究審批:此項審批適用於對利用中國人類遺傳資源開展國際合作科學研究的規範和管理。
國際合作科學研究備案:為獲得上市特許、國際合作、不涉及人類遺傳資源材料出境的。
人類遺傳資源材料出境審批
人類遺傳資源資訊對外提供或開放使用備案
保藏審批(很少)
申報流程?
如下圖所示。
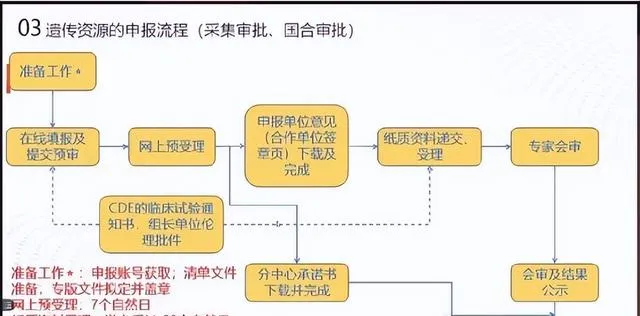
CRC需要配合完成什麽工作?
CRC需了解本中心遺傳辦承諾書蓋章流程、大約多久可獲得蓋章件。在取得本中心倫理批件後,配合CRA完成簽字、遞交等工作。
(5)協定簽署
協助CRA收集臨床試驗相關檢查檢驗費用。主協定由CRA根據醫院樣版或申辦方樣版擬定,CRC協定由SMO的PM根據醫院樣版或公司樣版擬定。公司、機構、雙方財務法務稽核完成後,一般由公司負責人先完成簽署,然後由CRC協助完成PI、科主任、機構簽署。各方簽署完成、機構蓋章後掃描一份電子版發送給CRA,ISF保存一份紙質版原件,其余交給各方保存。
(6)研究產品入庫
根據機構流程提前溝通好入庫時間、地點,按要求入庫儲存,註意不要超溫,保留運輸溫度記錄及研究產品交接單,定期匯出儲存溫度記錄。
(7)接收專案物資
與研究者協調物資、檔存放地點以及辦公地點。啟動前產生檔按ISF目錄歸檔。了解科室情況,預篩、儲備患者。
(8)票據管理
跟進倫理審查費用及研究者費發票。
2、專案啟動(啟動會)
(1)協助CRA了解中心啟動要求,如:首筆款到賬、啟動前質控等;
(2)與研究者預約啟動會時間,確定參會人員;
(3)啟動會當天協助布置會場,取餐分發等;
(4)完成專案方案培訓;
(5)協助研究者完成授權表、培訓表、啟動會簽到表、PDCF、FDF等表格簽字。
3、專案實施
(1)受試者管理
根據方案要求協助研究者進行受試者篩選、知情同意書簽署、入組、隨機分層,結束資訊的及時更新及受試者訪視日程安排等,同時嚴格按照方案入排標準督促入組合格受試者。及時收集受試者身份證、銀行卡資訊,準確填寫篩選入選表及鑒認程式碼表。督促受試者在家按要求填寫日記卡及問卷(如有)並按時返回研究中心。
(2)數據管理
按方案要求督促研究者評判化驗單、完善病歷,熟悉受試者既往病史病歷,協助準確記錄AE與合並用藥,協助填寫生命體征表(如有)、研究產品交接表、發放回收表(如有)、器械使用記錄表(如有);根據原始檔錄入數據、解答質疑,按申辦方節點配合數據清理;修改CRF時,用一條單線劃去錯誤的數據,保持清晰可辨,在不正確數據旁邊書寫正確數據,並簽署修改人姓名及日期,必要時備註修改原因,電子版 CRF 修改時,按照數據錄入系統要求修改。
知情同意相關名詞解釋:
受試者: 參與臨床試驗的個體,既可以是健康人也可以是患有特定疾病的患者。
知情同意: 是指向受試者告知一項試驗的各方面情況後,受試者自願確認其同意參加該項臨床試驗的過程,須以簽名和註明日期的知情同意書(Informed Consent Form)作為檔證明。
弱勢受試者: 指維護自身意願和權利的能力不足或者喪失的受試者,其自願參加臨床試驗的意願,有可能被試驗的預期獲益或者拒絕參加可能被報復而受到不正當影響。包括:研究者的學生和下級、申辦者的員工、軍人、犯人、無藥可救疾病的患者、處於危急狀況的患者,入住福利院的人、格拉斯哥流浪、未成年人和無能力知情同意的人等。
監護人: 指對無行為能力或限制行為能力的人身、財產和其他合法權益負有監督和保護責任的人。
公平見證人: 指與臨床試驗無關,不受臨床試驗相關人員不公正影響的個人,在受試者或者其監護人無閱讀能力時,作為公正的見證人,閱讀知情同意書和其他書面資料,並見證知情同意。

(3)研究產品管理
研究產品的接收、庫存清點、發放、回收、定期匯出並保存溫度記錄,庫存不足時及時提醒CRA進行申請,超溫時及時上報;
(4)生物樣本管理
樣本的處理、保存、清點、運送,定期匯出並保存溫度記錄,超溫及時上報;
(5)文件管理
定期整理ISF及受試者資料夾,檔產生後及時歸檔,以免遺失。
(6)SAE報告
發生SAE時,按申辦方上報流程,在研究者獲知的24小時內協助研究者上報至申辦方,並及時跟進受試者情況,完善隨訪報告。
(7)倫理遞交
根據中心要求及時協助研究者和申辦方CRA遞交各類更新檔至倫理、機構,包括修正案、年審、PD、SUSAR、DSUR、IB、研究產品檢測報告等,直至專案結束;
(8)配合
配合監查、稽查、檢查、質控,協助研究者進行問題解決及回復。
(9)票據管理
按中心流程進行受試者補貼的發放;跟進倫理審查費用及研究者費發票。
安全性事件定義:
不良事件: 是指在醫療器械臨床試驗過程中出現的不良醫學事件,無論是否與試驗醫療器械有關。
嚴重不良事件: 指受試者在臨床試驗過程中出現死亡、危及生命、永久或者嚴重的殘疾或者功能喪失、受試者需要住院治療或者延長住院時間,以及先天性異常或者出生缺陷等不良醫學事件。
可疑且非預期嚴重不良反應(SUSAR): 指臨床表現的性質和嚴重程度超出了試驗藥物研究者手冊、已上市藥品的說明書或者產品特性摘要等已有資料資訊的可疑並且非預期的嚴重不良反應。
器械缺陷: 是指臨床試驗過程中醫療器械在正常使用情況下存在可能危及人體健康和生命安全的不合理風險,如標簽錯誤、質素問題、故障等。
簡單來說不良事件分兩類,一類叫「事件」,諸如:不良事件、嚴重不良事件、重要不良事件,這類不一定與藥物有關;一類叫「反應」,如不良反應、副反應、非預期不良反應、非預期嚴重不良反應,這一類「反應」就和藥物一定是有相關性的。他們的關系如右圖,AE是所有安全性事件的總和,其中一部份是有關的,一部份是無關,在有關的和無關中,各有一部份是嚴重的,一部份是普通的,於是兩者的交集就是嚴重的不良反應,在嚴重的不良反應中有一部份是預期的,就是在開始研究前,我們已經知道的安全性資訊,一部份是非預期的,就是我們說的SUSAR(嚴重非預期不良反應)。
AE記錄要素:
1、AE的名稱: AE的名稱應該為醫學術語,應優先使用醫學診斷(可參考NCI-CTCAE 5.0)。AE也可以是癥狀、體征、實驗室異常、加重或新診斷的疾病等,在確定不良事件名稱時,應確保每個不良事件名稱由單一的事件組成,一個診斷、體征、癥狀就是一個不良事件。
2、持續時間: AE的開始時間、結束時間;
3、嚴重程度: 不良事件的分級標準應依據試驗方案所附的標準,常用的有 WHO,NCI-CTC AE或專業特定標準等。一般分為:輕、中、重或 NCI-CTCAE 1-5級。

4、與研究產品的關系: 國內常用的評判結果包括以下幾種
七分法:肯定、很可能、可能、 可疑、不相關、待評價、無法評價/判斷
六分法:肯定、很可能、可能、可疑、待評價、無法評價/判斷
五分法:肯定相關、很可能相關、可能相關、可能無關、不相關
二分法:相關、不相關
5、處理措施: 針對不良事件,是否處理、用藥、手術等,需記錄其處理方式;因不良事件對研究產品采取的措施,是否暫停或終止使用研究產品治療
6、轉歸: 恢復/痊愈、恢復/痊愈伴有後遺癥、好轉/緩解、未恢復/未緩解/持續、死亡、未知。(根據方案描述來記錄)
7、其他:
是否為嚴重不良事件(SAE),是否為AESI(特別關註的不良事件)等
4、中心關閉
(1)按中心要求完成結題質控並協助完成問題整改或問題回復;
(2)協助完成關中心前數據清理、鎖庫;
(3)協助完成研究產品及其他物資的清點、回收、銷毀;
(4)協助研究者倫理遞交結題審查;
(5)協助CRA進行尾款結算;
(6)按機構要求預約,完成試驗檔歸檔,臨床試驗必備檔應當至少保存臨床試驗結束後5年。











