來源:藥渡
撰文:幻目
突破性治療藥物
是指藥物臨床試驗期間,用於防治嚴重危及生命或者嚴重影響生存質素的疾病,且尚無有效防治手段或者與現有治療手段相比有足夠證據表明具有明顯臨床優勢的創新藥或者改良型新藥等。
為了適應不斷增長的藥物需求並跟上中國制藥領域的快速發展,為鼓勵創新和滿足臨床急需,2020年7月,國家藥監局釋出【突破性治療藥物審評工作程式(試行)】檔的公告,申請人可在Ⅰ、Ⅱ期臨床試驗階段,通常不晚於Ⅲ期臨床試驗開展前申請突破性治療藥物。
從CDE的公示資訊可以看出
2020年至今已有上百款藥物納入了突破性治療品種。藥品涵蓋了當下較為熱門的創新療法,如CAR-T療法、ADC、雙特異性抗體等。
本文就11月剛剛批準納入突破性治療藥物的品種進行整理,以此了解CDE對突破性治療藥物的要求,為企業申請突破性治療品種提供參考。
01
pCAR-19B
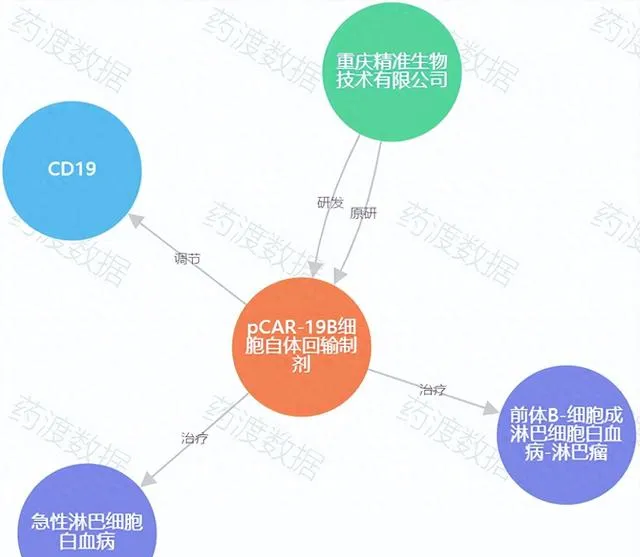
圖1. pCAR-19B知識圖譜,來源:藥渡數據
pCAR-19B是一款由重慶精準生物自主研發的人源化CD19 CAR-T,能夠顯著降低CAR-T細胞免疫原性,延長CAR-T細胞在體內的存續時間。據了解,pCAR-19B是由重慶精準生物采用其Precision CAR設計策略打造出的更為有效的CAR-T產品。pCAR-19B細胞自體回輸制劑已經於2019年獲兒童、青少年急性淋巴細胞白血病(B-ALL)適應癥註冊臨床默示特許。 它是國內首個針對該適應癥進入到II期註冊臨床的CAR-T產品,也是國內首個針對該適應癥的人源化設計的CAR-T產品。
該產品於2019年11月啟動I期註冊臨床試驗,納入9例符合標準的難治性或多線治療後復發的兒童或青少年B-ALL患者,且超過一半受試者為高瘤荷患者。數據顯示,9例患者均獲得完全緩解(CR),總體緩解率達100%,且首次達到完全緩解(CR)的患者微小殘留病變(MRD)也均為陰性;無劑量限制性毒性(DLT)和治療相關死亡事件發生,總體安全性和耐受性良好。2022年,pCAR-19B產品開始關鍵Ⅱ期註冊臨床試驗,該臨床試驗為多中心臨床試驗,目標入組人數為100人,主要目標為評價pCAR-19B細胞自體回輸制劑治療CD19陽性復發/難治性B細胞急性淋巴細胞白血病的3個月客觀緩解率(ORR)。
02
谷美替尼片
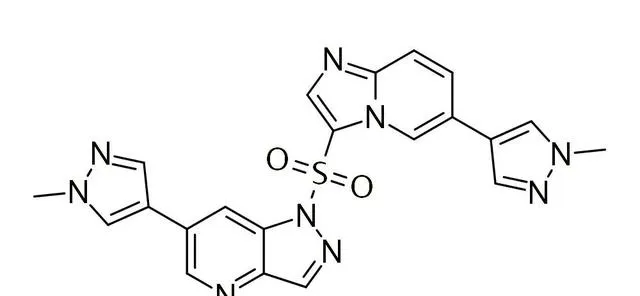
圖2. 谷美替尼化學結構式,來源:藥渡數據
谷美替尼(SCC244)是一種高選擇性c-Met抑制劑,該藥能夠選擇性抑制c-Met激酶活性,進而抑制腫瘤細胞的增殖、遷移和侵襲,在攜帶MET外顯子14跳躍突變的非小細胞肺癌患者中,表現出持久療效。臨床研究結果顯示,谷美替尼片具有優良的藥代動力學特性以及良好的安全性和耐受性,在人體中藥物半衰期長,穩態谷濃度高,有利於靶點的持續抑制。
據CDE網站顯示,該藥在2021年就曾納入了突破性治療品種。2023年3月,國家藥品監督管理局正式批準海益坦®(谷美替尼片)適用於治療具有間質-上皮轉化因子(MET)外顯子14跳變的局部晚期或轉移性非小細胞肺癌患者。
一項開放、國際多中心的單臂II期研究GLORY試驗(NCT04270591),主要評估了谷美替尼治療MET外顯子14跳變的局部晚期或轉移性NSCLC患者的有效性和安全性。在該試驗中,總共入組了84例患者,其中79例具有中心實驗室確認的MET外顯子14跳變的非小細胞肺癌患者,初治患者44例,經治患者35例。在所有人群中,由盲態獨立影像評估委員會(BIRC)評估的總體客觀緩解率(ORR)為66%。
谷美替尼起效迅速,絕大部份在首次腫瘤評估中達到緩解,中位起效時間為1.4個月。中位緩解持續時間(DOR)為8.3個月;中位無進展生存期(PFS)為8.5個月;中位總生存期(OS)為17.3個月;疾病控制率(DCR)為84%。在初治患者中ORR為71%,中位DOR為15.0個月;中位PFS為11.7個月;中位OS尚未達到。在經治患者中,ORR為60%,中位DOR為8.3個月;中位PFS為7.6個月。14例腦轉移患者中,有13例被納入療效分析集。
結果顯示,這些腦轉移患者的總體ORR高達85%,觀察到了令人鼓舞的顱內抗腫瘤作用。在安全性方面,整體安全可控。最常報告的治療相關不良事件包括水腫、低白蛋白血癥、頭痛、食欲不振和惡心,這些是接受MET抑制劑治療的患者中已知的常見不良反應。
03
BIVV020註射液
圖3. BIVV020知識圖譜 ,來源:藥渡數據
CIDP是一種罕見的自身免疫疾病,會影響到大腦和脊髓外的外周神經。罹患這種疾病的患者,其保護神經的髓鞘會出現損傷,導致肌肉出現麻木、虛弱、疲勞等癥狀。隨著病情的持續,CIDP的癥狀會進一步加重,極大地限制了患者的運動能力,並降低他們的生活質素。
BIVV020(SAR445088)是一種人源化IgG4單複制抗體,可與特定的絲胺酸蛋白酶 C1選擇性結合,從而阻止下遊酶級聯的啟用,導致C3轉化酶的啟用和膜攻擊復合物的形成。補體途徑也啟用促進巨噬細胞募集、炎癥和細胞裂解,同時使凝集素和旁路途徑在功能上保持完整,以便宿主防禦。在補體介導疾病的治療中,具有潛在的臨床益處。
其在93名健康參與者中進行了單次和多次給藥SAR445088的I期、首次人體雙盲、隨機、安慰劑對照、劑量遞增試驗,結果表明: SAR445088耐受性良好,在人類單次給藥和多次給藥後,均具有良好的PK和PD譜(藥代動力學和藥效學)。
這些發現支持對經典補體介導性疾病患者進行進一步的臨床研究。根據中國藥物臨床試驗登記與資訊公示平台官網,賽諾菲正在開展SAR445088治療慢性炎性脫髓鞘性多發性神經根神經病(CIDP)成人患者的II期概念驗證研究。該研究的主要目的為評價SAR445088在標準治療(SOC)經治、SOC難治和SOC未治人群三個CIDP患者亞群中的有效性,以及評價SAR445088在CIDP長期治療中的安全性和耐受性。
04
Vamorolone口服混懸液
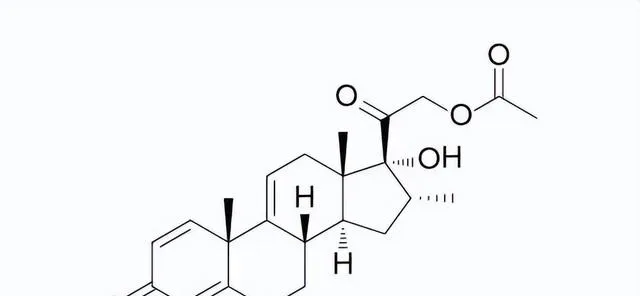
圖4. Vamorolone化學結構式,來源:藥渡數據
在杜氏肌營養不良癥(DMD)患者中,類固醇藥物的抗炎癥效果是介導藥物療效的主要機制,但是類固醇的其它活性會帶來影響患者生活質素的副作用。Vamorolone與普通類固醇藥物的區別在於——能夠有選擇性地啟用類固醇的某些訊號通路,能夠在保持類固醇對DMD療效的同時減少副作用的產生。
Vamorolone獲得美國與歐洲用以治療DMD的孤兒藥資格,並獲得美國FDA快速通道資格與罕見兒科疾病認定。2023年10月26日,美國FDA批準了Vamorolone口服混懸液40 mg/mL的上市申請,用於治療2歲及以上杜氏肌營養不良(DMD)患者。Vamorolone最初由ReveraGen BioPharma開發。2018年,Santhera公司獲得了該藥的全球研發特許(除日本和南韓以外)。2022年1月,曙方醫藥與Santhera公司宣布就vamorolone達成獨家授權協定,從而獲得該產品在大中華區用於DMD及其他罕見病適應癥的獨家開發和商業化權益。
FDA批準是基於一項關鍵2b期臨床試驗VISION-DM積極的臨床數據。試驗評估了vamorolone治療DMD患者的療效和安全性,結果達到了主要終點,試驗組在治療24周後由臥位至站立所需時間(TTSTAND,DMD兒童患者疾病惡化的第一項功能性指標)優於安慰劑組,且差異有統計學意義。另外,該研究許多次要終點的結果也與主要終點結果一致。Vamorolone在試驗中表現出良好的安全性和耐受性,最常見的不良事件是頭疼、嘔吐和維生素D缺乏,但不良事件的嚴重程度一般為輕至中度。
05
BRII-179
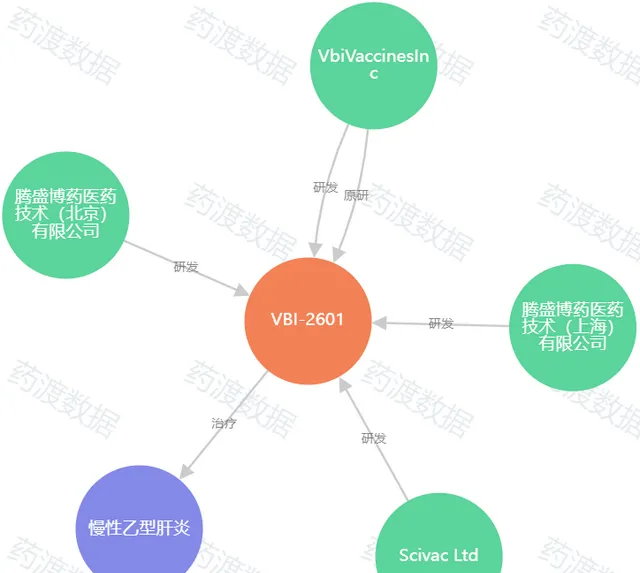
圖5. BRII-179知識圖譜 ,來源:藥渡數據
BRII-179是一種包含3種HBV表面抗原組成的治療性疫苗,作用機制為靶向增強B細胞和T細胞特異性免疫,從而實作慢性HBV感染功能性治愈的目標。2018年騰盛博藥從VBI Vaccines引進BRII-179,獲得了在大中華區的商業化權益。自2023年7月起,該公司還將BRII-179的獨家許可延伸至全球。
在現有PEG-IFNα(聚乙二醇幹擾素α)基礎上聯合BRII-179治療,總體安全且耐受性良好,其不良事件與既往報道的PEG-IFNα治療和BRII-179的不良事件相似。治療結束12周,與安慰劑+PEG-IFNα組相比,BRII-179+PEG-IFNα組的HBsAg(乙肝表面抗原)清除率更高。在治療結束24周仍觀察到該HBsAg清除率方面的差異,這種差異一直維持到第36周。臨床研究還發現,在第24周,聯合給藥組的HBsAg血清學轉換率顯著高於安慰劑+PEG-IFNα組。
06
恩沃利單抗
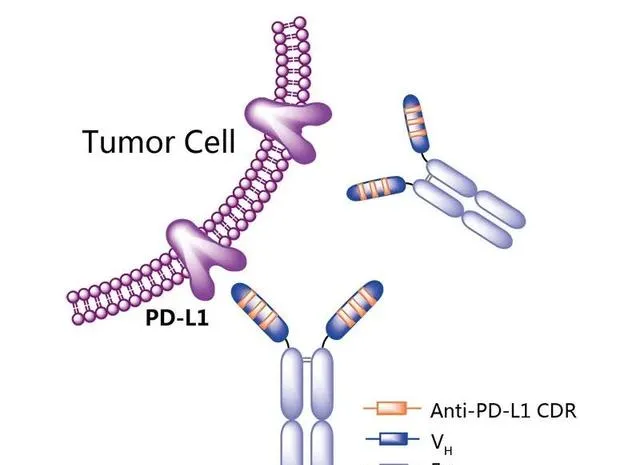
圖6. 恩沃利單抗作用機制示意圖,來源:藥渡數據
恩沃利單抗是一種由單域抗體(sdAb)和Fc段組成的單特異性抗體,分子量是完整抗體分子量的一半,這 使其具有增強的穿透性,同時具有完整的抗原結合能力。 此外,Fc-介導的效應功能在恩沃利單抗中被削弱,以限制其接觸免疫系統並避免意外的不必要免疫反應。2021年,NMPA批準上市適應癥為不可切除或轉移性微衛星高度不穩定(MSI-H)或錯配修復基因缺陷型(dMMR)的成人晚期實體瘤患者的治療。
侖伐替尼是一種口服多靶點激酶抑制劑,可選擇性抑制血管內皮生長因子(VEGF)受體的激酶活性,以及抑制其他促血管生成和致癌通路相關的受體酪胺酸激酶(RTK)活性,包括成纖維細胞生長因子(FGF)受體,血小板衍生生長因子(PDGF)受體,轉染重排(RET)等。免疫療法與抗血管生成靶向藥物聯用能顯著提高患者獲益。
早前,侖伐替尼原研產品已在美國獲批與PD-1抑制劑聯合用於治療特定晚期子宮內膜癌患者。而 恩沃利單抗與侖伐替尼聯用方案的優勢在於恩沃利單抗為皮下註射給藥,侖伐替尼為口服片劑,二者均用藥便捷,有利於提高患者依從性,實作腫瘤慢病化管理。 2023年10月,恩維達®聯合侖伐替尼治療復發性子宮內膜癌患者的Ⅲ期臨床研究(KN035-US-004)獲得FDA的新藥臨床試驗批準。
07
JMKX001899
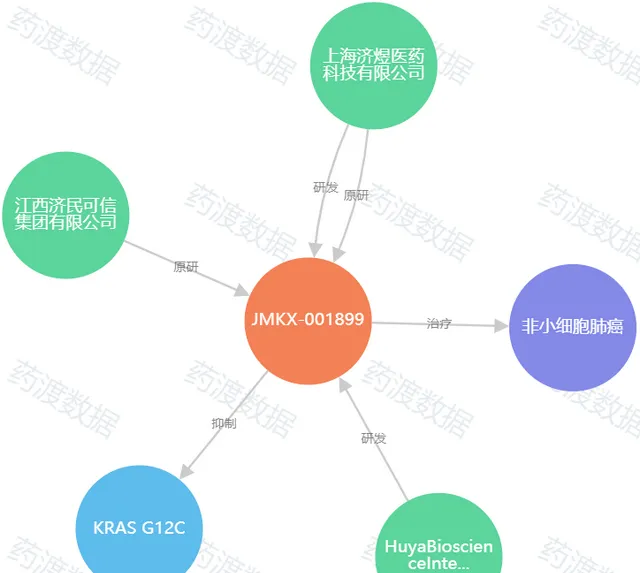
圖7. JMKX001899知識圖譜,來源:藥渡數據
JMKX001899是濟民可信集團研究院研發的一款口服新型小分子化合物,與KRAS G12C突變生成的半胱胺酸共價結合後,更傾向與鳥苷二磷酸(GDP)的結合,導致鳥苷三磷酸(GTP)與KRAS的親和力降低,透過將KRAS G12C突變體特異性不可逆地釘選在非啟用的GDP結合狀態,從而抑制腫瘤細胞增殖活性,達到抗腫瘤效果。
一項開放標簽、多中心 I/II 期臨床研究仍在進行之中,以評估JMKX001899片劑在攜帶KRAS G12C突變的晚期或轉移性實體瘤患者(Ia 期)和非小細胞肺癌患者中的安全性、耐受性、藥代動力學和抗腫瘤活性。
小結
CDE對納入突破性治療藥物審評程式的品種會采取一系列支持政策,有助於藥品走上「加速審批」的快車道,對於制藥公司是縮短研發時間、簡化試驗設計、提升公司知名度和融資的利器。但想要拿到「入場券」還需要藥物具有突破性的臨床價值。需要註意的是突破性治療僅針對創新藥或者改良型新藥,只針對嚴重危及生命和嚴重影響生存質素的兩類疾病,且臨床的安全有效性數據得到認可。
在目前內卷嚴重的制藥行業,申請突破性治療藥物或許將成為藥企的一條新出路。期待有更多的突破性治療藥品能加速上市,進一步滿足中國重大疾病的臨床治療需求,為無藥可醫的罕見病、腫瘤等患者帶來福音 。
*聲明:本文僅是介紹醫藥疾病領域研究進展或簡述研究概況或分享醫藥相關訊息,並非也不會進行治療或診斷方案推薦,也不對相關投資構成任何建議。內容如有疏漏,歡迎溝通指出!











