今天,美國FDA批準由Orchard Therapeutics所開發的基因療法Lenmeldy(atidarsagene autotemcel,arsa-cel)用於治療滿足特定條件的的異染性腦白質營養不良(MLD)兒童患者。
根據新聞稿,這是首個用以治療這項罕見遺傳疾病,且獲得FDA批準的基因療法。

MLD是一種罕見且危及生命的機體代謝系統遺傳性疾病
,根據現有文獻估計發生率約為1/10萬新生兒。MLD是由芳基硫酸酯酶-a(ARSA)基因突變引起的,導致硫酸鹽在腦和身體其他區域蓄積,包括肝臟、膽囊、腎臟和/或脾臟。隨著時間的推移,患者的神經系統會受損,導致其在運動、行為和認知上退化,並行生嚴重痙攣和癲癇發作等神經問題。MLD患者會逐漸喪失活動、說話、吞咽、進食和視物的能力。
在嬰兒晚期,發病後5年的死亡率估計為50%,10年的死亡率估計為44%。
Atidarsagene autotemcel(又名OTL-200)
使用慢病毒載體將編碼芳基硫酸酯酶-A的ARSA轉基因匯入到MLD患者的自體CD34陽性造血幹細胞和祖細胞中,以恢復芳基硫酸酯酶-A的表達,從而透過一次性治療,持續保留患者的運動功能和認知發育能力。
該療法已在歐盟獲批(商品名:Libmeldy)用於治療以ARSA基因雙等位基因突變導致兒童ARSA酶活性降低為特征的MLD患者。該藥物曾被美國FDA授予優先審評資格。
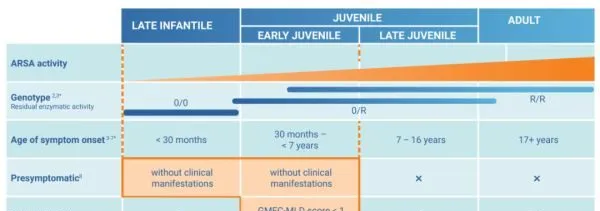
▲適合接受atidarsagene autotemcel治療的MLD患者群體(圖片來源:參考資料[3])
根據FDA新聞稿,該療法的安全性和有效性是根據37名兒童患者數據所進行評估,這些兒童在兩項單臂、開放標簽臨床試驗和一項擴大使用計劃中接受了atidarsagene autotemcel治療,所獲得的試驗結果與未接受治療MLD患者的自然史數據進行比較。試驗主要療效終點是無嚴重運動障礙生存期,定義為從出生到第一次出現失去支撐或無法坐立或死亡的時間間隔。
分析顯示,與未經治療的兒童相比,atidarsagene autotemcel治療顯著降低MLD兒童患者的嚴重運動障礙或死亡的風險。
所有接受治療的癥狀前晚期嬰兒形式的(pre-symptomatic late infantile)MLD兒童在6歲時均存活,而自然史組中只有58%的兒童存活。
5歲時,
71%接受治療的兒童能夠在沒有輔助的情況下行走。百分之八十五接受治療兒童的語言和表現智商得分正常,而未經治療的兒童尚未報告這一情況。
此外,患有癥狀前青少年早期形式(pre-symptomatic early juvenile)MLD和早期癥狀早期青少年形式(early symptomatic early juvenile)MLD的兒童表現出運動和/或認知疾病的減慢。
累積隨訪數據顯示,atidarsagene autotemcel治療通常顯示良好的耐受性,無治療相關嚴重不良事件或死亡。該療法最常見的副作用是發燒和白細胞計數低、口腔潰瘍、呼吸道感染、皮疹、病毒感染、發燒、胃腸道感染、肝臟腫大等。
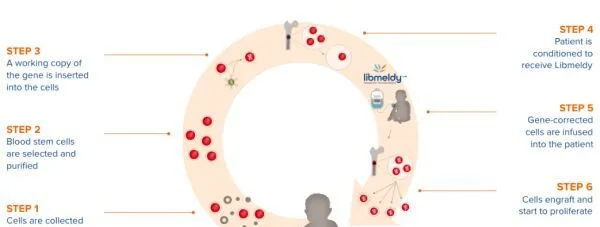
▲Atidarsagene autotemcel治療流程(圖片來源:參考資料[4])
去年10月,Kyowa Kirin和Orchard Therapeutics宣布,兩家公司達成最終協定,根據該協定,Kyowa Kirin將以大約3.874億美元收購Orchard公司。此交易為atidarsagene autotemcel的開發提供了更多資源。
目前,全球眾多醫藥企業正積極開發治療MLD的創新療法,許多臨床前潛在藥物有望在未來進入臨床試驗階段。
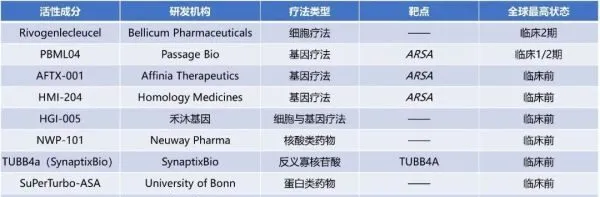
▲全球處於活躍研發狀態的MLD療法盤點(數據來源:公開資料,藥明康德內容團隊制圖)
註:本表由藥明康德內容團隊根據公開資料梳理,為不完全統計。如有遺漏,歡迎補充。











